虽然定制式
医疗器械可以通过豁免上市前审批得以生产、使用,但该类产品毕竟是少数,大多数产品仍然按照患者匹配器械通过审评审批流程进入市场流通。那么对于监管机构来说,需要建立技术审查指导原则,统一审评尺度,指导企业研发、生产和申报。
在这样的背景下,国家药监局医疗器械技术审评中心于2018年发布了《定制式增材制造
医疗器械技术审查指导原则(征求意见稿)》。后随着《定制式医疗器械监督管理规定(试行)》的发布,对该指导原则的名称和使用范围进行了修订,更名为《无源植入性骨、关节及口腔硬组织个性化增材制造医疗器械注册技术审查指导原则》,有望在今年正式发布。
该指导原则聚焦采用3D打印工艺制成的骨科、口腔个性化医疗器械的技术审评关注点,首次按照医工交互条件和医工交互能力两方面,把技术审评关注的要点串联起来。医工交互条件是对软、硬件的客观要求,包括个性化设计和增材制造生产工艺整个流程中的软件、3D打印设备、原材料、打印工艺、后处理工艺、验证测试。而医工交互能力是指由医生和工程师组成的团队的设计开发整体实力,由于个性化医疗器械的个性化设计、生产和使用,医工合作是贯穿始终的。如果设计开发能按照医工交互条件和能力的要求完成,已经能够基本保证医疗器械的安全性和有效性,所以该指导原则的覆盖范围比较全面。
出于对未知领域的尊重和留白,该指导原则增加了适用范围的限定。但是未知问题有时可以采用惯常模式和已知方法解答。比如,定制式医疗器械虽然采用备案制,无需技术审评,但是生产企业仍然需要制定一套完善的质量管理体系,保证每批次产品的质量可控,那么本指导原则的相关内容可以作为参考。
虽然监管部门已经制定并发布了针对个性化医疗器械的法规和指南,搭起了监管框架,但是,个性化医疗器械监管中还存在许多管理和技术层面的问题和挑战,需要面对与解决完善。关于个性化3D打印医疗器械的一系列指导原则正在陆续制定中,对此,我们充满期待。
 吸引器
吸引器 高频电刀
高频电刀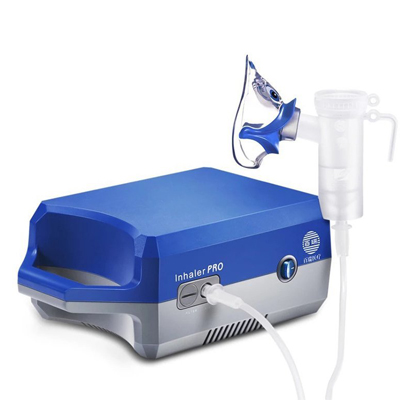 雾化器
雾化器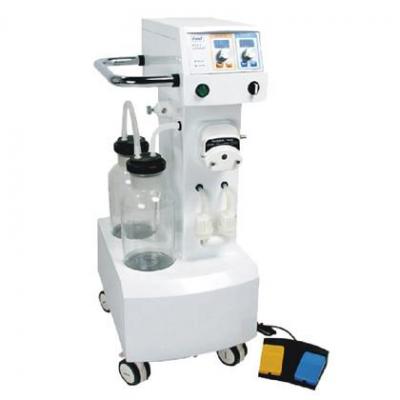 吸脂机
吸脂机 洗胃机
洗胃机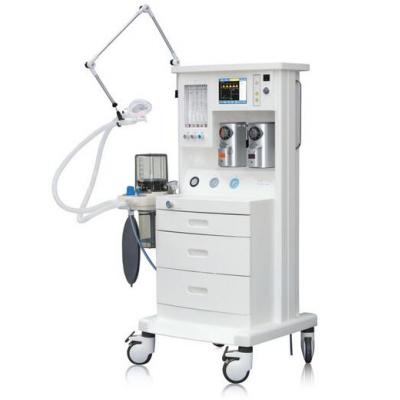 麻醉机
麻醉机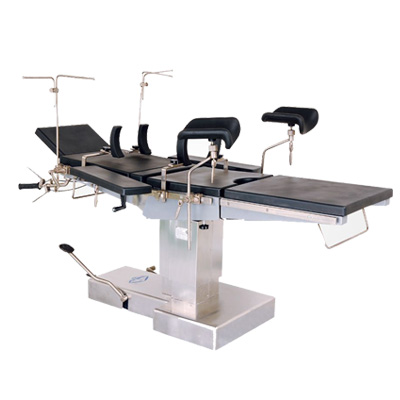 手术台
手术台 无影灯
无影灯
 400-006-1850
400-006-1850 沪公网安备 31011502008791号
沪公网安备 31011502008791号